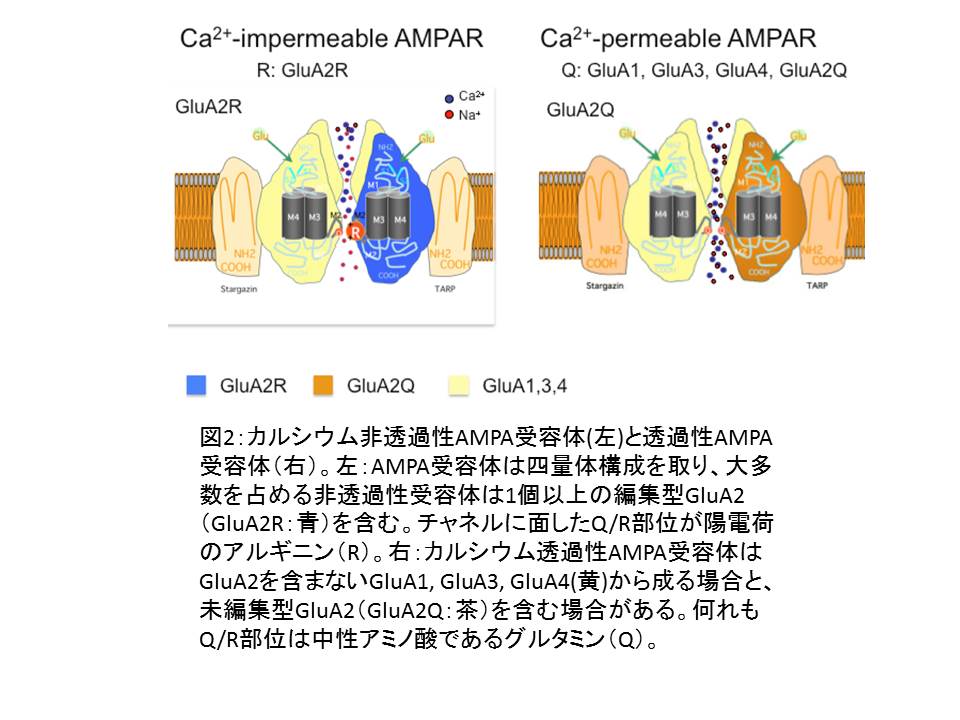
 (図2)
(図2)(ア) AMPA受容体(グルタミン酸受容体サブタイプ
① グルタミン酸受容体はイオンチャネル型と代謝調節型がある。AMPA受容体はNMDA受容体、カイニン酸受容体などと共に前者に属す。
② AMPA受容体は速い神経伝達を制御する神経細胞の興奮性を規定するグルタミン酸受容体で、中枢神経に広く分布している。
③ AMPA受容体は4種類のサブユニット(GluA1~ GluA4)の様々な組み合わせに依る四量体構造をとり、Na+/ K+を透過させることで、膜電位を制御している。(図2)
④ サブユニット構成によりCa2+ 透過性のものと非透過性のものがある(図2)。
Ca2+非透過性AMPA受容体が圧倒的大多数を占める。Ca2+ 透過性AMPA受容体の過剰発現は動物にてんかんを引き起こすことが知られていた。
(イ) GluA2サブユニットとそのRNA編集(RNA editing) (図2)
① AMPA受容体と
Ca2+ 透過性
1. AMPA受容体のCa2+透過性はGluA2がサブユニットに含まれるかどうかで決まる。正常のニューロンが発現するGluA2は全て編集型(②で後述)である。
2. Ca2+非透過性AMPA受容体:ニューロンに発現するAMPA受容体の大多数を占め、編集型GluA2 をサブユニットに含む
3. Ca2+透過性AMPA受容体:サブユニット構成にGluA2を欠くもの(生理的には海馬の抑制性ニューロンやグリア細胞)と、未編集型GluA2を含むもの(生理的にはグリア細胞の一部に存在するとされる)とがある。
② 未編集型GluA2の発現の意味するもの
1. GluA2はグルタミン・アルギニン部位(Q/R部位)でRNA編集[1]を受ける。
2. すなわち、GluA2 pre-mRNAエクソン11にあるグルタミン(Q)コドン(CAG)のアデノシンがイノシンに置換され、翻訳時にイノシンはグアノシンとして認識されるので、アルギニン(R)(CGG
= CIG)に翻訳される。ゲノム情報CAGがタンパクレベルではR (CGG)に置換される。
3. Q/R部位はチャネルに面しており、陽荷電のRはCa2+ の透過をブロックするが、中性のQはブロック出来ない。
4. AMPA受容体の他の3種類のサブタイプのQ/R部位にはRNA編集が起こらず、Q型のため、GluA2をサブユニットに持たないAMPA受容体やGluA2がサブユニットに含まれていても未編集の場合、AMPA受容体のCa2+ 透過性は高い。
5. ヒトを含む哺乳類ではGluA2のQ/R部位は100%編集され、GluA2の発現もAMPA受容体サブユニットの過半を占めるため、ニューロンに発現するAMPA受容体は殆どがCa2+非透過性である。
6. GluA2Q/R 部位にRNA編集が起こらないことで
a)AMPA受容体のチャネル特性が変わる(Ca2+透過性になる)
b)未編集型GluA2は編集型GluA2に優先してシナプス後膜へ輸送される
c)そのため、未編集型GluA2が発現した分、機能的AMPA受容体に占めるCa2+透過性AMPA受容体の割合が増す。(ニューロンへのCa2+流入は未編集型GluA2が100%になると野生型の29倍、50%でも4〜5倍とされている。7
d)GluA2のRNA編集が出来ない変異動物はてんかん重積のため、生後3週以内に死亡してしまう。8,9
7. したがって、未編集型GluA2の発現自体が少量であっても異常である。
(ウ)RNA編集酵素ADAR2 (adenosine deaminase acting on RNA 2)
 (図3)
(図3)
① 哺乳類の中枢神経ではRNA編集が極めて盛んに行われている。その大多数はアデノシン・イノシン(A-I)置換である。
② このA-I置換は二重鎖RNAに働く酵素により行われ、哺乳類では3種類のアイソフォーム(ADAR1-ADAR3)が知られている。
③ GluA2 Q/R部位のA-I置換は特異的にADAR2により触媒され、ADAR2の欠損ではこの部位のRNA編集は起こらない。
④ ヒトでの検討でADAR1,ADAR2とも中枢神経では主にニューロンに発現し、A-I置換に関與しているが、ADAR3は
中枢神経特異的な発現パターンを持ちながら、発現は主としてグリアで、A-I置換活性も明らかでない。10
(ア) 孤発性ALSでは、運動ニューロン選択的に、未編集型GluA2が発現している11,12。
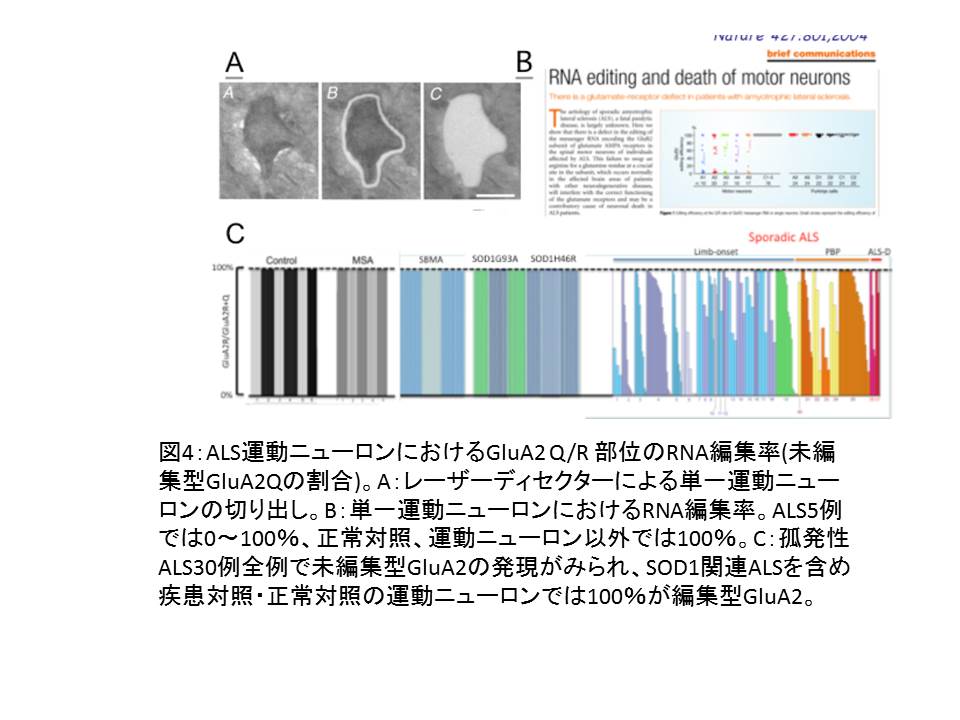 (図4))
(図4))
① 単一運動ニューロンに発現する未編集型GluA2の発現割合は、GluA2 mRNA全体の0%~100%6と大きくばらつく。11
② GluA2 mRNAの発現は低下していない14。
③ 運動ニューロンに未編集型GluA2が発現しているALS症例でも、小脳のプルキンエ細胞では発現していない(部位選択的) 11。
④ 30例以上の孤発例全例で未編集型GluA2を発現する運動ニューロンがみられる(汎発性)14。
⑤ 他の運動ニューロン疾患 SODI関連ALS(ALS1)、球脊髄性筋萎縮症(SBMA)を含む疾患対照、健常対照の運動ニューロンは、すべて編集型GluA2のみを発現している
(疾患特異性)11,12,15。
(イ) 孤発性ALSの運動ニューロンではADAR2の発現低下に依るA-I置換活性が低下している。
 (図5)
(図5)
① ADAR2 mRNAの発現レベルの低下、ADAR2特異的RNA編集部位における未編集型mRNAの割合の増加2,14。
② ADAR1、ADAR3 mRNA発現量、ADAR1特異的編集部位のRNA編集活性には変化がない14。
③ 未編集型GluA2を発現する孤発性ALS運動ニューロンのADAR2 mRNA発現レベル低下は著しいが(正常対照の1/100)、編集型GluA2のみを発現する運動ニューロンでも既に低下している(正常対照の2/3~1/2)14。
(ウ) 以上は我々が見出した異常だが、RNA編集異常の発見の後、TDP-43病理(後述)も疾患特異性の高い病態であることが日米の研究グループから独立に報告された。16,17
3. ADAR2低下に依る未編集型GluA2発現の意味:分子病態モデルマウス(コンディショナルADAR2ノックアウトマウス;ADAR2flox/flox/VChAT-Cre.Fast; AR2マウス)を開発し、運動ニューロン死の原因かどうかを解析を用いた解析(図6)
(ア) 運動ニューロン死の原因であることの証明
① ADAR2缺損マウスはけいれん重積のため生後三週以内に死亡してしまう。運動ニューロン死が起こるかどうかは不明だった。
② そのため、運動ニューロン選択的にADAR2を欠損させたコンディショナルADAR2ノックアウトマウス(AR2)を作成18。
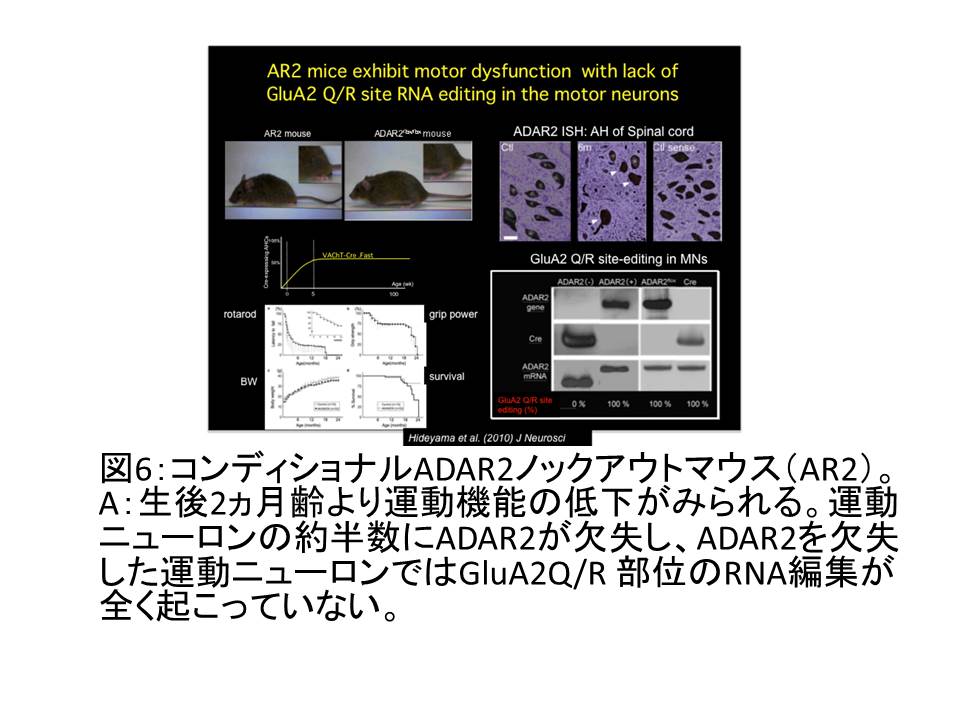 (図6A)
(図6A)
③ AR2マウスでは 18
1. 進行性の運動機能低下がみられる(図6A)。
2. 約半数の運動ニューロンではADAR2がCre依存性にノックアウトされ、その運動ニューロンではGluA2のRNA編集が起こらない(図6A)
3. ADAR2を缺損した運動ニューロンはすべて細胞死に陥る。変性の速度は25%〜50%/月程度で、残存するADAR2を缺損した運動ニューロン数はF(t)=A(1/2)bt(t:時間、A,b:係数)に近似する。神経変性疾患に特有な「緩徐進行性の神経細胞死」を引き起こす。
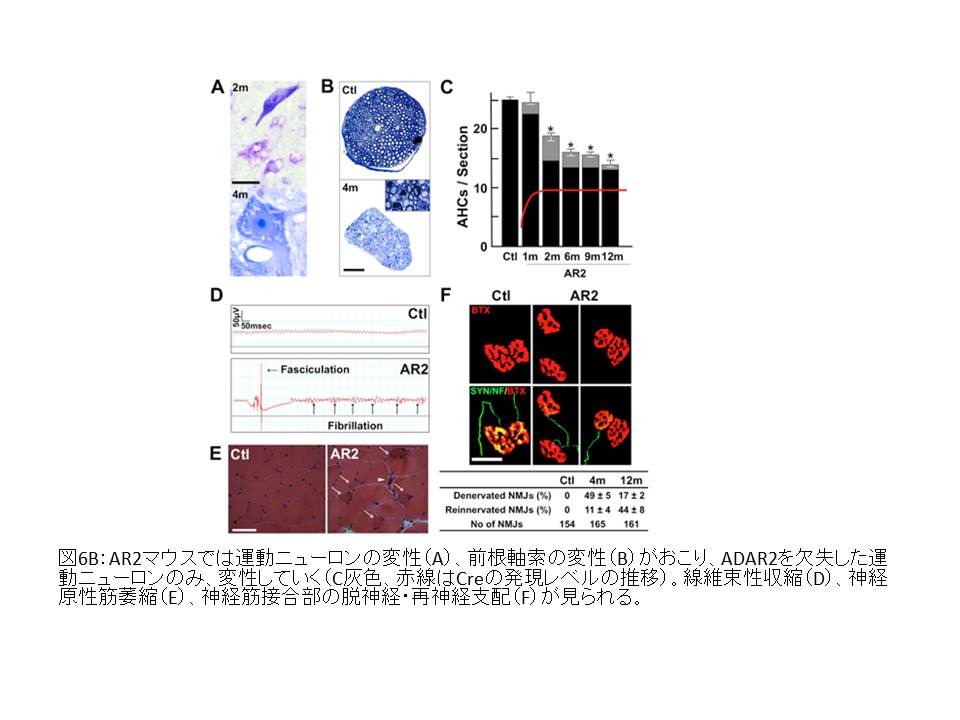 (図6B)
(図6B)
④ 運動ニューロンの変性は、遺伝子に編集型GluA2を組み込んだ変異マウスとの掛け合わせにより阻止される。
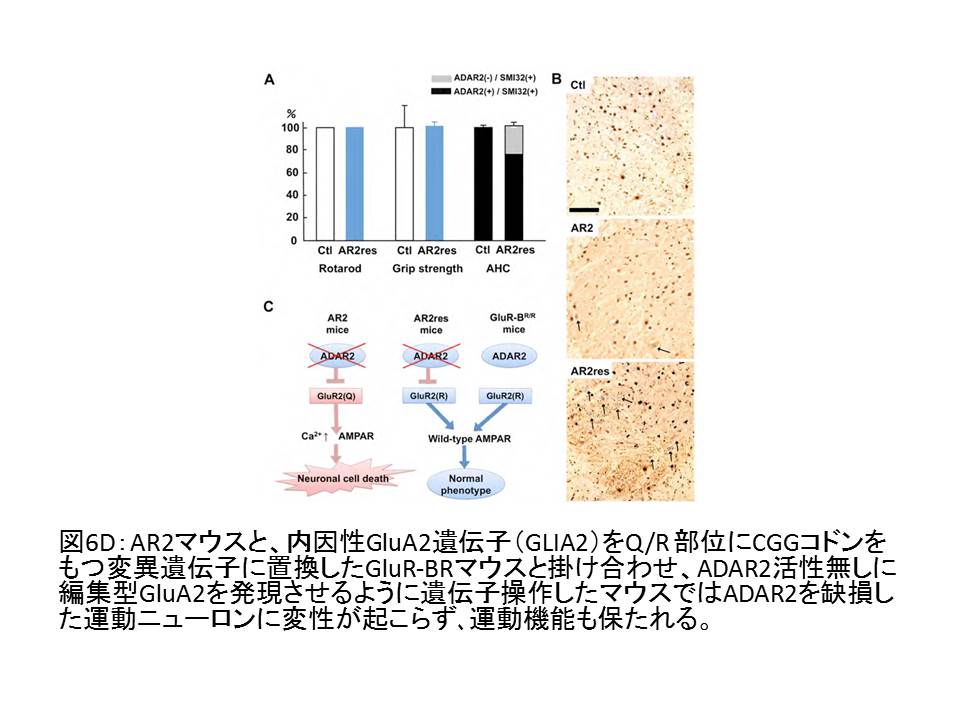 (図6D)
(図6D)
このマウスはADAR2の缺損した運動ニューロンでも編集型GluA2を発現するので、ADAR2の発現低下そのものではなく、未編集型GluA2発現が細胞死に直結する分子異常であることを示している18。
(イ) ALS相同の運動ニューロンにおける細胞死の選択性
① ALS同様、外眼筋ニューロンには未編集型GluA2が発現しているにもかかわらず脱落が見られない18。
 (図6C)。
(図6C)。
② 外眼筋ニューロンが細胞内Ca2+濃度上昇に抵抗性である理由としての仮説。
1. 細胞内Ca2+バッファータンパクの発現が運動ニューロンより遙かに高い
2. 豊富な抑制性(GABA作動性)ニューロンの入力がある
(ウ) 運動ニューロンの生存にとり必要なADAR2 発現レベルは正常の50%以上
① ADAR2発現低下と未編集型GluA2発現
1. (ア)−④に記載したように編集型GluA2の発現自体が運動ニューロンにとり有害なので、全てのGluA2のQ/R 部位を編集するのに必要なレベルのADAR2 発現が必要である。
2. ヘテロ接合体AR2マウスADAR2+/flox/VChAT-Cre.Fast; AR2H)では、運動ニューロンの2割が未編集型GluA2を発現し、同数の運動ニューロンが脱落する。AR2HではADAR2アリルの片方の発現は保たれ、約50%のADAR2活性と考えられるので、ADAR2発現の半減はALS発症の危険水域といえる。19。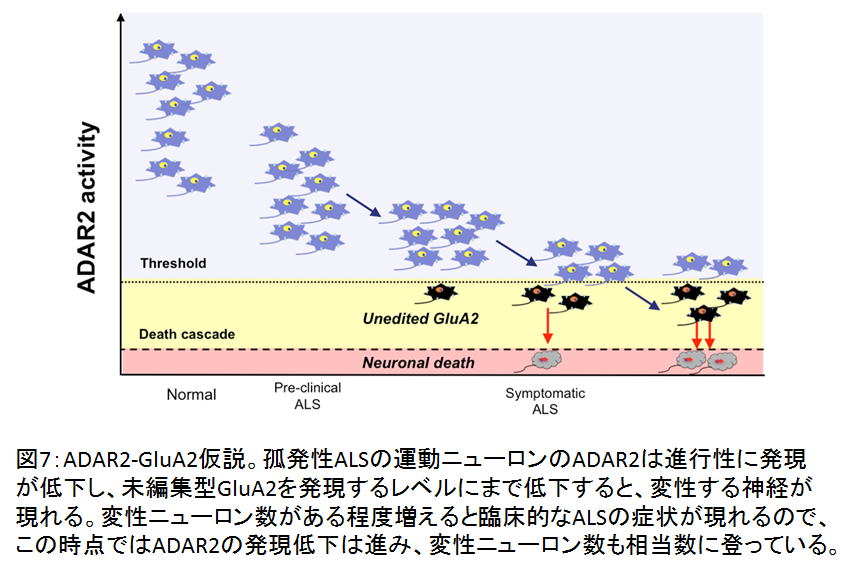 (図7)
(図7)
3. ALS患者の剖検脊髄から切り出した単一運動ニューロン組織の解析から、編集型GluA2のみを発現している運動ニューロンではADAR2 mRNAの発現レベル低下は正常対照の2/3〜1/2程度(すなわちADAR2 活性が50%以下になると未編集型GluA2 が発現する)
② 未編集型GluA2発現とカルシウム透過性AMPA受容体発現
1. 1−(イ)−②に記載したように、Ca2+を過剰に透過するAMPA受容体が発現する。そのため、運動ニューロン内のCa2+濃度が異常に上昇する。
③ このような研究結果から、ALSの病因にはADAR2発現低下に依る未編集型GluA2の発現が大きな役割を果たしているという仮説を提唱している19。
(図7)
(ア) TDP-43病理:ALS患者脊髄運動ニューロンにみられる神経病理学的指標
① TDP-43病理:本来は核に局在するRNA結合タンパクであるTDP-43が、ALSの運動ニューロンでは核から喪失し、細胞質に異常な封入体が形成されている。しかも、TDP-43は異常な断片化、リン酸化を受けている。
② 大多数の孤発性ALS、一部の遺伝性ALS患者で運動ニューロンにTDP-43病理が観察されることから、ALSの神経病理学的指標となっている。
③ 孤発性ALS運動ニューロンでは、TDP-43陽性封入体の出現とADAR2の免疫活性消失とが同一の運動ニューロンに生じており、TDP-43陽性封入体を持たない
運動ニューロンではADAR2免疫活性が保たれている20。
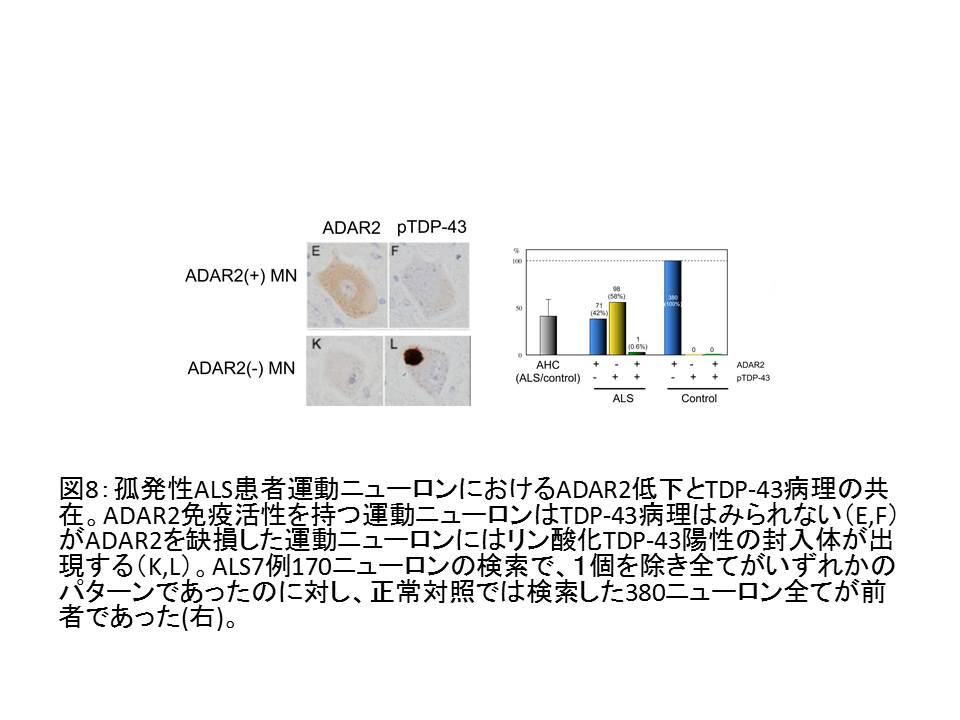 (図8)
(図8)
④ すなわちTDP-43病理が出現メカニズムとADAR2が低下するメカニズムの間には何らかの連関がある20。
(イ) 分子病態モデルマウス(AR2マウス)における所見
① ADAR2低下とTDP-43病理の分子連関の検討を行い、ADAR2活性低下がTDP-43病理を引き起こすことを明らかにした。
1. 培養細胞での検討からTDP-43の断片化、過剰発現、ノックダウンによる減少、変異TDP-43の発現は何れも培養細胞のADAR2 発現低下を引き起こさない21
2. 分子病態モデルマウス(AR2マウス)のADAR2 を欠損した運動ニューロンでTDP-43病理類似の細胞内局在異常が観察される22
3. そのメカニズムは、AMPA受容体から流入した過剰なカルシウムがタンパク分解酵素であるカルパインを活性化し、TDP-43を凝集しやすい断片に切断することであることを証明した22
② カルパイン断片に相同の断片がALS患者剖検脳・脊髄にも検出された22 ことは、ALSの運動ニューロンでも同様のメカニズムによりADAR2 活性低下がTDP-43病理を引き起こしている可能性を示唆する。
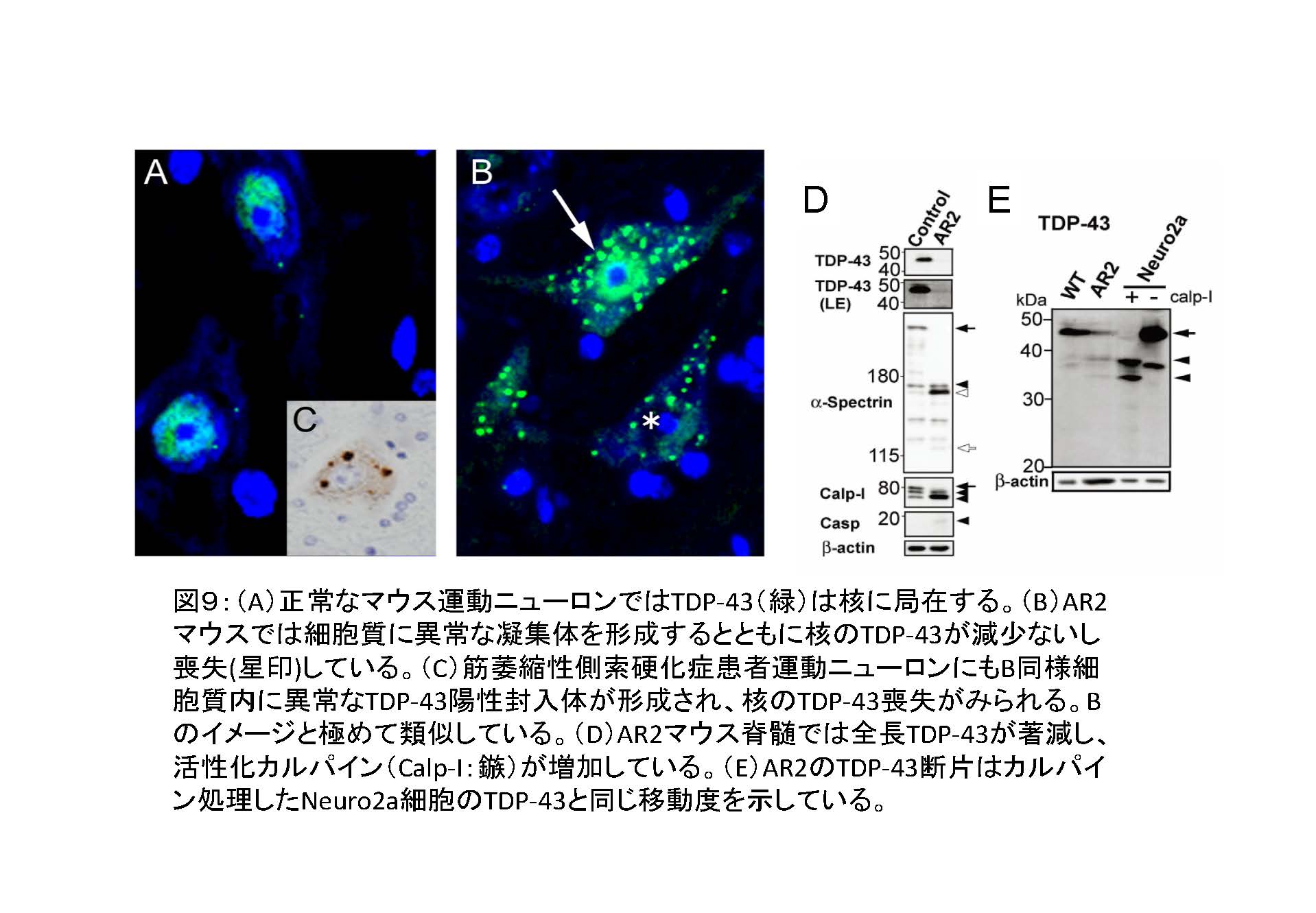 (図9)
(図9)
(ウ) ADAR2 活性低下に依りTDP-43病理が形成されるメカニズム
ADAR2 発現低下 → Q/R 部位未編集型GluA2の発現→Ca2+透過性AMPA受容体の発現 → 細胞内Ca2+濃度の上昇 → Ca2+依存性プロテアーゼであるカルパインの活性化 → TDP-43の断片化→断片の凝集性昂進による凝集塊・封入体形成 → 核と細胞質をシャトルするTDP-43の封入体への巻き込み → 核からのTDP-43の喪失。
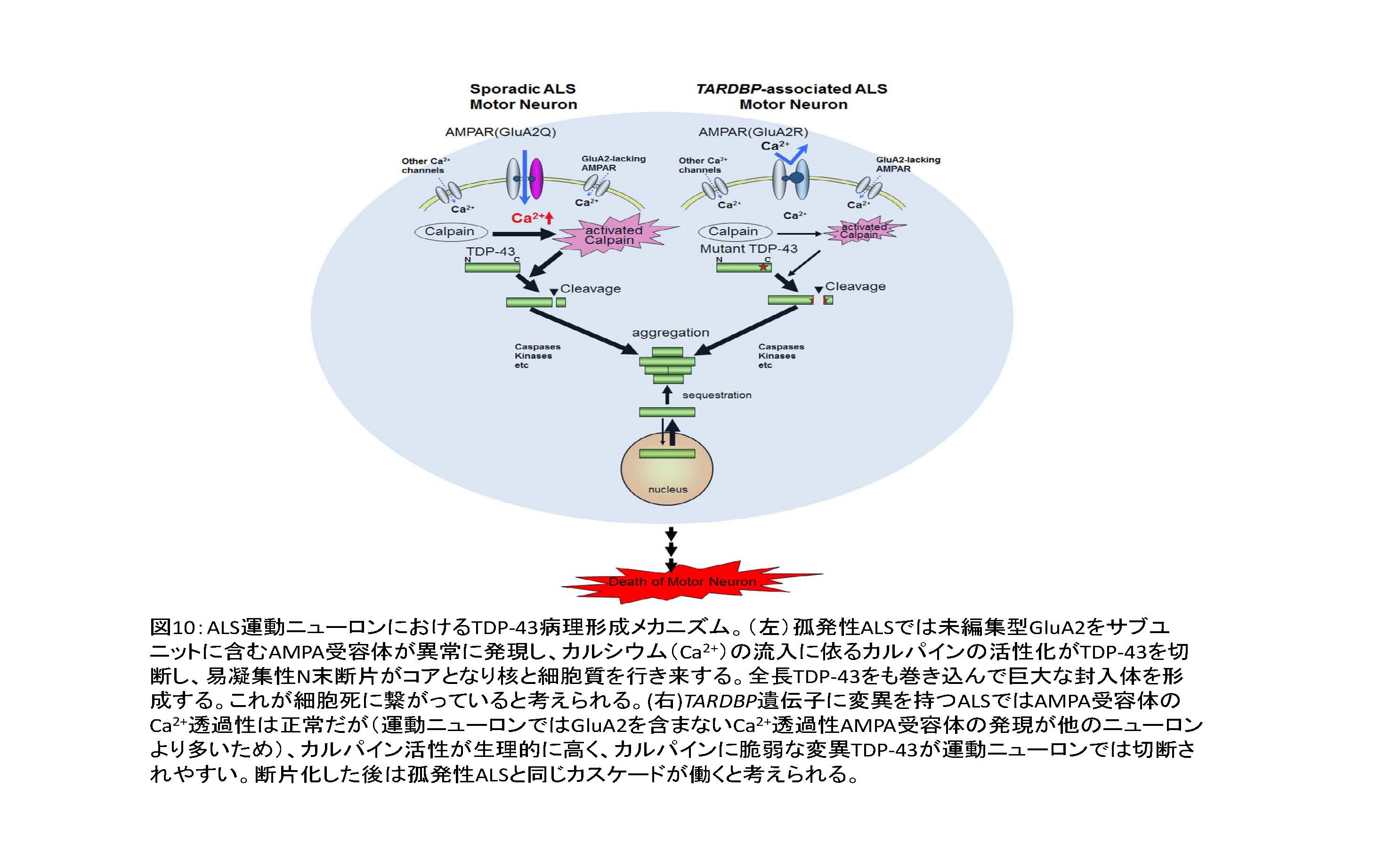 (図10左)
(図10左)
(エ) 孤発性ALS以外のTDP-43病理
① 一部の遺伝性ALSの運動ニューロンにもTDP-43病理が観察される。
1. TARDBP(TDP-43をコードする遺伝子)関連ALS
2. C90RF72関連ALS
② 大脳皮質や海馬ニューロンなどにTDP-43病理が観察される
1. 前頭側頭葉変性症、アルツハイマー病、拳闘家脳症など
2. 正常高齢者
(オ) TDP-43病理の形成にはカルパインが中心的に関與していることが明らかになったので、これら孤発性ALSのTDP-43病理形成にどのようにカルパインが関わるかが今後の検討の対象になる。
① カルパインの活性化は細胞内Ca2+濃度の上昇によるので、複数の細胞内Ca2+濃度を高めるシステムのどれが働くかにより、異なったメカニズムでカルパインが活性化しうる。
1. NMDA受容体、Ca2+チャネルなど細胞外からの流入
2. ミトコンドリアや小胞体など細胞内のCa2+貯蔵庫からの放出
② カルパイン活性が高すぎると、易凝集性TDP-43断片はより小さい可溶性の断片に切断される。
1. TDP-43病理の形成には、ほどよいカルパインの活性が持続する細胞内環境が必要
2. 脳虚血や脳外傷ではNMDA受容体などからの大量のCa2+流入があり、しかも一過性なので、TDP-43の局在異常が一過性であり、封入体形成を伴わない
22
(図11)
3. TDP-43のリン酸化はカルパインによる切断活性への抵抗性を増強する23
(カ) カルパインの活性化以外に、TDP-43のカルパインへの脆弱性が増すことによっても、易凝集性断片が生じうる。
① TARDBP(TDP-43をコードする遺伝子)に変異のあるALSは、変異TDP-43がカルパインに切れやすくなることにより起こる。
② 運動ニューロンにはAMPA受容体のカルシウム透過性を決定するGluA2の発現が他のニューロンに比べ低いため、GluA2をサブユニットにもたないAMPA受容体、すなわちCa2+透過性AMPA受容体の発現が多く、活性型カルパインの発現も多いことがTARDBPの変異がALSとして現れる理由であろう22
(図10右)、図12
(キ) カルパインの活性化とTDP-43病理形成との関連は、超高齢マウス運動ニューロンにも生じている。
(図13)
① 加齢と共に運動ニューロン脱落が生ずること、加齢がALSの危険因子であることはよく知られており、その理由はよくわかっていなかった。
② 運動ニューロン(特にFast fatigable motor neuron)では加齢性にADAR2の発現が低下する24。加齢がALSの危険因子であることの分子基盤であると考えるとこの現象をよく説明できる。
③ 加齢は神経変性疾患の危険因子であることが多い。運動ニューロン以外でも加齢と共にTDP-43病理が観察されるようになるので、加齢依存性の細胞内Ca2+上昇がさまざまな神経疾患に病因的意義を持つ可能性がある。
(ア) 核の異常
① AR2マウス運動ニューロンの核にはCa2+の流入依存性に大きな空胞ができている26→ 核膜に異常があるだろう、この異常は細胞死に繋がる可能性がある
② 正常な核膜は核と細胞質の分子移動を制限しており物質の通過は核膜孔複合体Nuclear Pore Complex (NPC)を通じて行われる。
1. 核-細胞質の物質輸送:核→細胞質(RNA)、細胞質→核(タンパク RBP)
2. 輸送システム:NPC、Importin、Exportin、など
(イ) AR2マウス運動ニューロンの核膜異常26
(図14)
① NPCが破壊されている:Nupsの免疫活性消失
② 核・細胞質輸送に関わる分子の異常:Importin、Exportinの免疫活性消失
③ 核膜の構造的破壊:Lamin B(核の裏打ちタンパク)の消失
(ウ) カルパインがNups(NPC構成タンパク)を切断
① Nupsはカルパインにより特異的に分解される
② (イ)はカルパインが活性化したADAR2 欠損運動ニューロンに観察される
③ ADAR2活性低下に依るAMPA受容体からのCa2+流入増大がその原因。TDP-43病理の原因でもある(前項(エ))
(エ)核・細胞質物質輸送の破綻と細胞死
① 運動ニューロンでの転写活性低下
1. リン酸化RNA Polymerase IIの発現している運動ニューロン数が減少
② 運動ニューロンの生存に必要な遺伝子発現が低下=細胞死の原因
(オ)ALSの運動ニューロン26
① 孤発性ALSではAR2マウスと同様のNPC変化がみられる
② NPCの異常はTDP-43病理と共存している
③ すなわち、ADAR2 活性が低下し、カルパインが活性化している運動ニューロンに観察される
④ C9ORF72関連ALSでも核-細胞質の物質輸送障害が病因的意義を持つことが報告されている
(カ)これらのことから、ADAR2発現低下→未編集型GluA2発現→Ca2+透過性AMPA受容体発現→カルパインの活性化→TDP-43病理、と平行して
① →NPC破綻→遺伝子発現変化→運動ニューロンの生存に必須な遺伝子発現の低下→緩徐な細胞死と言う分子カスケードが考えられる。
(図15)
(キ)ADAR2 発現低下、未編集型GluA2の発現は孤発性ALSのみならず、一部の遺伝性ALSの運動ニューロンにも運動ニューロンにも確認されている
① FUS関連ALS:FUSP525L変異27
(ク)ADAR2発現低下の分子メカニズムを解析している。
6.分子病態の解析に基づいた孤発性ALSの分子治療へ
(ア)
(イ)
① ADAR2 活性の賦活
1. 期待される効果は、未編集型GluA2の発現抑制、AMPA受容体のCa2+透過性の正常化、それによる運動ニューロン死の阻止
a)薬剤によるADAR2活性上昇30
b) 遺伝子導入によるADAR2発現上昇
② 未編集型GluA2を含むAMPA受容体の特異的阻害
1. 期待される効果は、AMPA受容体からのCa2+流入阻害により細胞内Ca2+濃度上昇の抑制
a) 薬剤
b) 低分子化合物
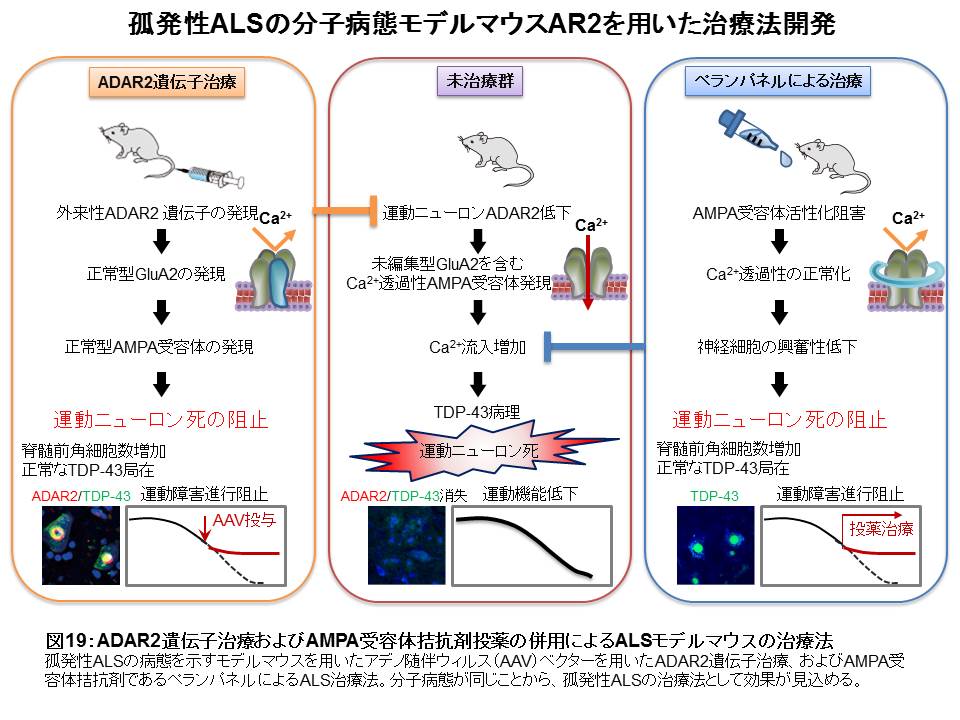
(ウ)これまでに行った前臨床治療研究成果
① ADAR2 活性の賦活
1. 培養細胞を用いたADAR2 賦活活性薬剤のスクリーニング
a)複数のADAR2 活性賦活物質が得られている
b)問題点としては、in vivoでの効果が必ずしも得られない、効果が持続しない、など
2. 分子病態モデルマウスを用いたADAR2 遺伝子による遺伝子治療。31
(図16)
a)ヒトADAR2 遺伝子(ADARB1)を搭載したAAV9ベクターを経静脈的ルートによりモデルマウスに投与
b)モデルマウスに一回投与することにより、運動ニューロンでADAR2 の発現レベルを1.5倍に増加させた
c)発症後投与でも、運動機能障害、運動ニューロン死を阻止し、TDP-43の局在異常を正常化した(図16)
d)ニューロン特異的プロモーターを用いることにより、末梢臓器での発現は見られず、中枢神経においても、ADAR2 遺伝子発現によるグリア細胞の増勢などの副作用は全く見られなかったことから、重篤な副作用は生じないと考えられる
② 未編集型GluA2を含むAMPA受容体の特異的阻害
1. 分子病態モデルマウスを用いたAMPA受容体阻害剤による治療32
(図17)
(図18)
1. Kwak, S., Hideyama, T., Yamashita, T. & Aizawa, H. AMPA receptor-mediated neuronal death in sporadic ALS. Neuropathology 30, 182-188 (2010).