特に言語発達遅滞を認める(75%が発語なし、25%は単語のみ、コミュニケーションはジェスチャーが主となる)
行動異常(50%)自傷行為、かんしゃく、社会的交流が苦手
少数例には髄鞘化遅延、脳室周囲結節性異所性灰白質の報告あり
そのうちの1/3は薬剤抵抗性
1p36欠失症候群とは?
【概念】
1番染色体、1p36領域の欠失が原因でおこる染色体微細欠失症候群。
特徴的な顔貌、筋緊張低下、精神発達遅滞、言語発達遅滞を認める。
【頻度】
5000〜10000人に一人と推定されている。男:女=1:2と、女児のほうが多い。
【臨床症状】 (Battaglia and Shaffer, GeneReviews 2008より抜粋)
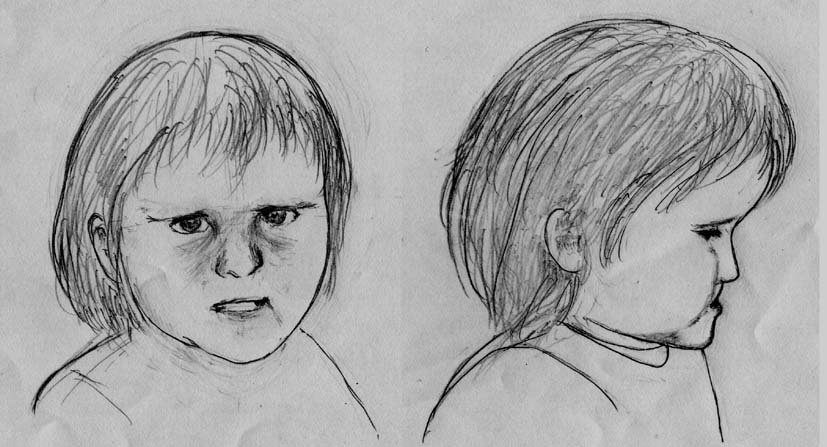
| 特徴的な顔貌(全例) | 小頭症、短頭症、直線状眉毛、落ち窪んだ目、内眼角贅皮、顔中央低形成、幅広い鼻根部、長い人中、小顎症、耳介低位 |
| 精神発達遅滞(全例) | 90%が重度、10%は軽度〜中等度 特に言語発達遅滞を認める(75%が発語なし、25%は単語のみ、コミュニケーションはジェスチャーが主となる) 行動異常(50%)自傷行為、かんしゃく、社会的交流が苦手 |
| 筋緊張低下(95%) | 嚥下障害(72%)、哺乳不良 |
| 頭部構造異常(88%) | 脳室拡大、くも膜嚢胞、大脳皮質低形成、脳梁低形成 少数例には髄鞘化遅延、脳室周囲結節性異所性灰白質の報告あり |
| 大泉門の閉鎖遅滞(77%) | |
| 心奇形(71%) | 種類は様々(心房中隔欠損、心室中隔欠損、動脈管開存、ファロー四徴症、大動脈狭窄、心筋緻密化障害) |
| てんかん(44-58%) | 初発は4day~2y、20%は早期乳児てんかん性脳症(EIEE)を認め、 そのうちの1/3は薬剤抵抗性 |
| 眼科的異常(52%) | 斜視、眼振、屈折障害 |
| 難聴(47%) | ほとんどが感音性のタイプ |
| 骨格の異常(41%) | 骨年齢遅延、側彎、肋骨異常 |
| 外性器の異常(25%) | 停留精巣、陰嚢低形成、外性器低形成、子宮低形成 |
| 腎奇形 (22%) | 水腎症、腎転位 |
| 甲状腺機能低下(15〜20%) |
【鑑別疾患】
Prader-Willi症候群、Angelman症候群、Smith-Magenis症候群
【染色体欠失パターン】
| 欠失 | 52% |
| 中間部欠失 | 29% |
| 複雑な構造異常を含む(欠失と重複、inverted duplication deletionなど) | 12% |
| 不均衡型転座による欠失 | 7% |

G-band法では大きい範囲の欠失(>5Mb)、不均衡型転座の診断は可能である
FISH法では端部欠失の有無は確認できるが、1と3の区別はできない。
アレイCGH法では1〜4すべてを正確に診断できる。
検査の流れ:Gband法→FISH法→アレイCGH法
欠失範囲は、共通のブレークポイントが存在しているわけではない。
【治療、予後】
リハビリや、療育指導が中心となる。
サインでのコミュニケーションの獲得
医療的なフォローとしてはてんかんの内服治療、栄養のサポート、
所見を認めた場合には循環器科、眼科、耳鼻科、整形外科でのフォロー
【遺伝カウンセリング】
次子へのリスクは、欠失のパターンが4種類のうちのどれかによって異なってくる。
欠失領域と臨床症状との関連性は、まだ明らかになっていないところもある。